Day :
- Track 7:Case Reports on Pathology
Session Introduction
Shadab Shireen
Bombay Hospital Institute of Medical sciences and Research Centre, India
Title: Diamond-gardner syndrome

Biography:
Dr. Shadab Shireen is pursuing her MD in Pathology in Bombay Hospital Institute of Medical sciences and Research Centre, Mumbai, India. She has done one international publication. Interested in research work.
Abstract:
Diamond-Gardner Syndrome or autoerythocyte sensitization is a rare syndrome characterised by spontaneous development of painful edematous skin lesions progressing to ecchymosis over the next 24 hours. Severe stress and emotional trauma always precede the skin lesion. It is regarded primarily as an autoimmune vasculopathy with sensitization to phosphatidyl-serine, a component of erythrocyte stroma.
We present here a case of 15 years old girl who presented with multiple ecchymotic patches over body. Baseline biochemical, hematological and immunological investigations were normal. Skin biopsy showed no evidence of vasculitis. All routine coagulation investigations were normal. Diagnosis of Gardner-Diamond syndrome was made clinicaly, it was therefore diagnosis of exclusion. A high index of suspicion was necessary to make the diagnosis.
References: 1.Gardner FH, Diamond LK. Autoerythrocyte sensitization: A form of purpura producing painful
bruising following autosensitization to red cells in certain women. Blood. 1955;10:675–90.
2. Ogston D, Ogston WD, Bennett NB. Psychogenic purpura. Br Med J. 1971;1:30.
3. Hanna WT, Fitzpatrick R, Krauss S, Machado E, Dunn CD. Psychogenic purpura
(autoerythrocyte sensitization) South Med J. 1981;74:538–42.
4. Yücel B, Kiziltan E, Aktan M. Dissociative identity disorder presenting with psychogenic
purpura. Psychosomatics. 2000;41:279–81.
5.James WD, Berger TG, Elston DM. 10th ed. Philadelphia: Saunders Elsevier; 2006. Andrew's
diseases of the skin.
6. Anderson JE, De Goff W, McNamara M. Autoerythrocyte sensitization (psychogenic
purpura): A case report and review of the literature. Pediatr Emerg Care. 1999;15:47–8.
CASE REPORT:
A young female 15 years of age presented with recurrent ecchymotc painful patch on body since last 4 months, it started with feeling of severe pain and burning in skin on anterior part of right thigh then eventually appearance of red patch over that area few hours later which was painful associated with severe muscular pain, reached to size of 7×4cm and became swollen, progressed to become blue with yellowish hue next 48 hours, due to pain patient was unable to move her lower limb, with time it became less tender and gradually disappeared in 1 week and involved other parts of the body later. On examination largest patch was on right forearm, it was bluish in color, swollen, tender and measured 4×3cm. There was associated carpo-pedal spasm of right hand. During the stay in hospital patient had multiple altrernating somatic complaints like pain in Temporo-mandibular joint,locked jaw,
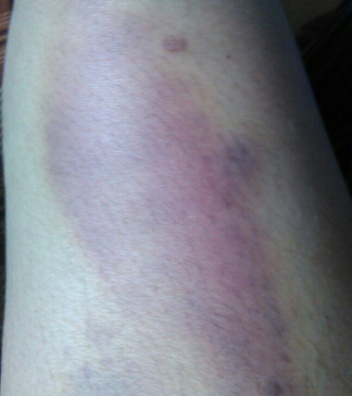
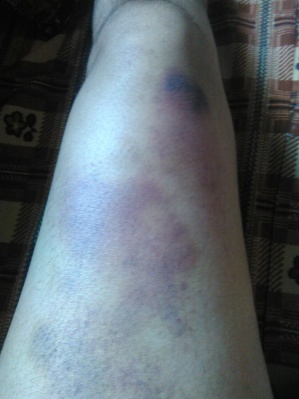
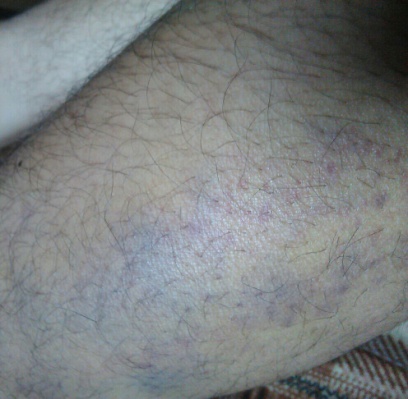
(A) (B) (C)
FIG.A, B, C: Showing echhymotic patches at different stages of development.
head ache, abdominal pain, pain in lower limbs with dorsiflexion of feet.
There was no history of use of aspirin, NSAIDS, heparin, warfarin and steroids. And no family history of bleeding disorder.
Psychiatric evaluation and tactful history taking revealed that the patient is having anxious and introvert personality, presently in stress to get good score in her exams and her social interaction has decreased as compared to past. One episode of ecchymosis started just before her exams suggesting possibility of stress as a precipitating cause of illness. Moreover patients alternating somatic complaints were relieved on distraction which was regarded as conversion disorder.
General and systemic examinations were non-contributory. Treatment with steroids for symptomatic relief showed no improvement.
Battery of lab tests including CBC, platelet studies, ANA, ANCA, RF, Anti-CCP,APLA, Anti-TPO Ab,C3, C4, TSH, Prolactin, IgG, Biochemistry, routine coagulation tests were carried out which helped to rule out ITP, VonWillibrand’s disease, DIC, cellulitis , SLE , Polyarteritis nodosa, Henoch-Schonlein purpura. Histopathological examination of skin biopsy was inconclusive.
Histories taken from family members regarding history of stress and psychiatric evaluation as well as Dermatology, Hematology and Psychiatric consultation helped us in diagnosing the patient as a case of ‘Gardner-Diamond Syndrome’. The diagnosis was made clinically. It was therefore ‘diagnosis of exclusion’ and high index of suspicion was required to make the diagnosis.
Clinical diagnosis is made difficult owning to the obscure nature of the disease, absence of standard laboratory markers and the unreliable nature of many patients histories. Which in turn increase patient morbidity, stress and cost on investigations.
Gardner-Diamond Syndrome is a rare condition that should be consider in the differential diagnosis of conditions in which unexplained eccymoses or purpuric lesions are noticed without any deranged lab investigations. Pschyological evaluation is of vital importance to decrease unnecessary investigations and incorporation of appropriate therapy. Psychotherapy and psychiatric treatment provides the most effective treatment to relieve the triggering stress factor.
Deniz Arik
Eskisehir Osmangazi University, Turkey
Title: Water-clear cell adenoma of the mediastinal parathyroid gland

Biography:
Deniz ARIK has completed his PhD at the age of 24 years from Hacettepe University and postdoctoral studies from Ankara Numune Research and Teaching Hospital. He has published more than 20 papers in reputed journals.
Abstract:
Water-clear cell adenoma is composed of cells with abundant clear-pink cytoplasm. It is postulated that the water-clear cells are transformed from the chief cells. We report a mediastinal water-clear cell adenoma of the parathyroid gland. At microscopic evaluation, uniform, large clear cells with fine cytoplasmic vacuolization, without nuclear atypia, arranged in solid and acinar patterns were revealed. The cells form nests separated by fine fibrovascular septae and stain positively with anti-parathyroid hormone. Primary hyperparathyroidism is characterized by the autonomous production of parathyroid hormone resulting in hypercalcemia. It is most commonly seen with sporadic adenomas, followed by hyperplasia, multiple adenomas, and carcinoma. Water-clear cell parathyroid adenoma is extremely rare. There have only 16 cases were reported in the English literature. To the best of our knowledge this has not been reported previously in this localisation. In differential diagnosis of clear cell lesions of the mediastinum, WCCA should be kept in mind.
- Track 13: Endocrinology Case Reports
Location: Duai, UAE
Session Introduction
Jamal Abdulkarim
Warwick medical school, UK
Title: Pancreatic Variant Anatomy: The ‘V shaped Pancreas’ – a case report

Biography:
Dr. Abdulkarim had completed his postgraduate training in radiology in Leicester training scheme UK where he obtained the FRCR. Currently he is a Consultant Radiologist at George Eliot Hospital and a visiting fellow to Warwick medical school. Dr Abdulkarim’s current research interest is in the field of reduction of intravenous contrast in CT examination and the effects on renal function where he had published several papers.
Abstract:
ABSTRACT:
Recognition of pancreatic anomalies on imaging is essential as they may be of clinical relevance and are potential causes of recurrent pancreatitis or gastric outlet obstruction in patients. (1)
We describe a ‘V shaped pancreas’, a pancreatic anomaly that resembles canine pancreatic anatomy that, to the best of our knowledge, has not previously been described in humans.
We will also review pancreatic embryological development and anatomy, including common variants.
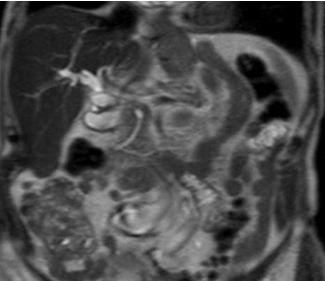
Fig 1: T2 weighted turbo spin echo single shot coronal sequence showing 2 separate moieties of pancreatic tissue separated by fat.
References:
1-Türkvatan A, Erden A, TürkoÄŸlu MA, Yener Ö. Congenital Variants and Anomalies of the Pancreas and Pancreatic Duct: Imaging by Magnetic Resonance Cholangiopancreaticography and Multidetector Computed Tomography. Korean Journal of Radiology 2013;14(6):905-913 doi:10.3348/kjr.2013.14.6.905.
2-Douglas H. Slatter, Textbook of Small Animal Surgery, 2003 - Veterinary surgery
3-Schulte SJ. Embryology, normal variation, and congenital anomalies of the pancreas. In: Stevenson GW, Freeny PC, Margulis AR, Burhenne HJ, editors. Margulis' and Burhenne's alimentary tract radiology. 5th ed. St. Louis: Mosby; 1994. pp. 1039–1051.
4-TaylorAJ, Mortele K. Congenital disorders of the bile and pancreatic ducts [abstr]. In: Abdominal Radiology Course annual meeting program. San Antonio, Tex, 2005; 224–226.
5-Koenraad J. Mortelé, Tatiana C. Rocha, Jonathan L. Streeter, and Andrew J. Taylor. Multimodality Imaging of Pancreatic and Biliary Congenital Anomalies RadioGraphics 2006 26:3 , 715-731
Anas Alghamdi
King Saud Bin Abdul-Aziz University for Health Sciences,Saudi Arabia
Title: Slipped capital femoral epiphysis and primary hyperparathyroidism: A case report

Biography:
Anas A. Alghamdi has completed the Bachelor's of Medicine from college of medicine of King Saud University at Riyadh, Saudi Arabia. Membership of Saudi Diabetes & Endocrine Association. Currently on postgraduation residency training program at King Fahad Medical City.
Abstract:
The aim of reporting this case is to highlight the association of two disorders: primary hyperparathyroidism and slipped capital femoral epiphysis. They are usually seen in two different age groups and rarely together. Primary hyperparathyroidism is a rare cause of slipped capital femoral epiphysis and only ten cases have been reported in the literature worldwide. The patient in our report is a 13-year-old girl who presented to our clinic with bilateral knee pain and a waddling gait. Subsequent investigations showed that she had primary hyperparathyroidism and slipped capital femoral epiphysis with low bone mass. On admission, a parathyroidectomy was performed; then, the slipped femoral epiphyses were fixed with satisfactory results. A systematic algorithmic approach that was illustrated in a previously published case was used. Such cases should be managed in a systematic approach based on the patient’s clinical status to prevent future morbidity. A literature review was conducted by performing a Medline search of all reported cases of primary hyperparathyroidism and slipped capital femoral epiphyses.

Biography:
Dr. Shyam Krishnan has completed his MBBS from B.J Medical College, Pune and Post-graduation in Pulmonary Medicine from Government Medical College, Surat, India.He is currently pursing his fellowship in Interventional Pulmonology at Fortis Hospital, Kolkata, India.
Abstract:
A 44 year old male transferred to our facility from another hospital with a diagnosis of an asthma exacerbation which was not responding to treatment (iv steroids/bronchodilators /antibiotics). He has had 2 admissions in the past 3 months for asthma exacerbations. His complains of shortness of breath on exertion associated with wheezing, nocturnal awakening and dry cough started 6 months ago. Patient denied chest pain, weight loss, fever, chills, night sweats or hemoptysis. No significant past medical history. He was a non-smoker. No history of allergies, nasal discharge, skin disorders. Treatment with bronchodilators did not alleviate symptoms.
On examination the patient was clinically stable. A fixed monophonic wheeze could be heard over the trachea and lung fields. Blood investigations were normal. Pulmonary function studies demonstrated a moderate obstructive pattern with no significant bronchodilator response. Despite a normal chest radiograph the fixed monophonic wheeze on auscultation prompted us to do a CT scan thorax to rule out any endo bronchial pathology. CT Scan demonstrated a polypoid intra-tracheal lesion occupying a 2 cm length of trachea. There was no mediastinal extension or adenopathy. Flexible fiber-optic Bronchoscopy was done which revealed a polypoid intra-tracheal mass occluding approximately 80% of the lumen. The tumor was removed in three sessions by Flexible Bronchoscopy with Argon plasma coagulation and cryosurgery. Histopathological and immunohistochemistry analysis of the resected tumor revealed an intra-tracheal schwannoma. The patient was put on bronchoscopic surveillance for one year with no evidence of recurrence. He is currently asymptomatic.
This case reinforces the common adage that “all that wheezes is not asthma”. Apart from unravelling a rare diagnosis (intra-tracheal schwannoma) the therapeutic role of the bronchoscopic ablative procedures for treatment of intra-tracheal masses are highlighted. More importantly as a result of these advances this patient was spared a major cardiothoracic intervention.
Maswood Ahmad
King Fahad Medical City, Riyadh
Title: An interesting case of neurobrucellosis presenting like pseudotumorcerebri.

Biography:
Dr. Maswood, currently persuing Fellowship in Endocrinology and Metabolism at Obesity, Endocrine and Metabolism Center, King Fahad Medical City, Riyadh. He is M.D. in Internal Medicine from India, actively involved in research recently and have published 5 papers. His feild of interest is Thyroid and General Endocrinology.
Abstract:
Thyrotoxic periodic paralysis (TPP) is a potentially lethal complication of hyperthyroidism characterized by recurrent muscle weakness and hypokalemia. It has been commonly reported in non-Asian populations. Four cases were reported in Saudis so far, and one had a life-threatening arrhythmia. We describe an additional case of a 28-year-old apparently healthy Saudi male patient, who presented with acute paraparesis associated with hypokalemia (K: 2.0 mmol/L), complicated by ventricular tachycardia and cardiac arrest. He was successfully resuscitated and his hypokalemia was corrected. A diagnosis of Graves’ disease associated with TPP was made. He was initially treated with carbimazole and β-blockers and then given a definitive therapy with radioactive iodine, which showed a good response. This case highlights the importance of early recognition and prompt treatment of TPP as a differential diagnosis for muscle weakness. A brief review of TPP and associated arrhythmia is included.
Nawaf Alhazmi
College of Medicine, University of Hail, Saudi Arabia
Title: Relationship between glycated hemoglobin and macrominerals in renal dialysis patients of Hail, Saudi Arabia

Biography:
Nawaf Oudah M Alhazmi is a medical student at College of Medicine, University of Hail, Hail, Kingdom of Saudi Arabia.
Abstract:
Essential minerals have significant role in the glucose metabolism and energy production inside the cell. Imperfect minerals metabolism have been associated with the increased mortality of renal dialysis patients, but their effects in these patients are less characterized. The literature suggested that the incidence of renal dialysis patients in Saudi Arabia showed rapid increase over the last 3 decades. In the present study, we examined the correlations between levels of minerals (serum calcium, phosphorus and magnesium) and HbA1c in diabetic and non-diabetic renal dialysis patients of Hail region. Total 76 blood samples of renal dialysis patients (diabetic and non-diabetic) were analyzed by using biochemical methods. As expected, no significant relationship was observed (p>0.05) in baseline parameters such as age, sodium, potassium, bilirubin, creatinine, urea and glucose, in both diabetic and non-diabetic renal dialysis patients. The results also showed that there is no significant relationship between calcium and phosphorus, calcium and magnesium as well as magnesium and phosphorus in non-diabetic renal dialysis patients; however, in diabetic patients calcium and phosphorus have minor significant association (p=0.057). Further, there was no significant relationship between phosphorus and HbA1c in both types of renal dialysis patients. However, in diabetic renal dialysis patients there was significant relationship (p<0.05) between calcium and HbA1c as well as magnesium and HbA1c. These preliminary results indicate the supportive role of calcium, magnesium and Hb1Ac in the better management of diabetes. The supplementation of calcium and magnesium might be beneficial to manage energy level associated with weakness in the diabetic patients.
- Track 3:Case Reports on Obstetrics & Gynecology
Session Introduction
Badreldeen Ahmed
Feto maternal medicine Centre, Qatar
Title: A case of massive fetal neck tumor

Biography:
Professor Badreldeen is the Director of the Feto-Maternal Medicine Centre in Doha and Professor of Obstetrics at Weill Cornell Medical College – Qatar. He previously held the position of Chairman of Obstetrics and Gynecology at Women’s Hospital, part of Hamad Medical Corporation. Professor Badreldeen was also a consultant obstetrician and gynecologist at Dorset County hospital in Dorchester, UK. His main interests are fetal medicine and high risk pregnancy. Professor Badreldeen is the founding member of the international society of Ultrasound in Obstetrics and Gynecology (ISUOG), a board member of the World Association of Prenatal Medicine, founder and president of the African Association of Perinatal Medicine, founder of the fellowship program in advanced training in obstetrics and gynecology, board member of the Fetus as the Patient Society, founding member of the Academy of Medical Educators and the Regional Director of the Ian Donald School of Medical Ultrasound. Professor Badreldeen has also published over 50 papers in peer-reviewed journals and is the Editor of the ‘Basic book of Ultrasound in Obstetrics and Gynecology’. He has written many chapters in several books, acts as a reviewer for many international journals.
Abstract:
Patient name: R. M,Age: 24
Parity: Para 4 SNVD. Presentation: At 28 weeks of gestation with a neck mass, no other obvious
Anomaly, no polyhydramnios
The mass in the fetal neck is partially solid partially cystic and minimal vascularity, diagnosis of fetal neck teratom was made with US and supported by Antenatal MRI.
Patient was booked for Ex Uterine Intrapartum Treatment (Exit procedure). This procedure will
Offers the advantage of insuring uteroplacental gas exchange While on Placental support.
This is complicated procedure require sophisticated synchronized team work involving multiple disciplines which include:
Fetal medicine specialist, pediatric surgeon, pediatric ENT and neonatologists.
The case management involves different specialties for a better outcome.

Biography:
Dr. Spandan Chaudhary is team leader of Medical Genetics, Diagnostics and Next Generation research divisions of Xcelris Labs, India. He has six years of professional experience in genomics industry specifically in the medical genetics segment. He has developed more than 30 very important diagnostic, sports and nutrition health related assays. He has designed the beta thalassemia mutation screening assay based on whole gene sequencing of HBB gene including all the mutations and indels. He has screened more than 200 individual samples and 25 trio samples as prenatal screening for beta thalassemia. This approach is very useful in diagnosing prenatal thalassemia in combination with regular screening methods
Abstract:
β-Thalassemia is a genetic disease characterized by reduced or non-functionality of β-globin gene expression, which is caused due to number of genetic variations and indels (insertions and deletions). In the present case study, we have reported a rare occurrence of compound heterozygosity of two different variants, namely, HBBc.92G>C and HBBc.92+5G>C in maternal amniotic fluid sample. Prenatal β-thalassemia mutation was detected using nucleotide sequencing method. After analysis, the father was found to be heterozygous for HBBc.92G>C (Codon 30 (G>C)) mutation (β0 type) and the mother was heterozygous for HBBc.92+5GNC (IVS I-5 (G>C)) mutation (β+ type). When amniotic fluid sample was analyzed for β- globin gene (HBB), we found the occurrence of heterozygous allelic pattern for aforesaid mutations. This compound heterozygous state of fetus sample was considered as β+/β0 category of β thalassemia which was clinically and genotypically interpreted as β-thalassemia major. The probability of occurrence of both mutations is very low, because mutations are only 5 base pairs apart on HBB gene. Segregation of compound heterozygosity has occurred twice in this family. Along with the present case, we will share our experience of analyzing 21 unrelated families (trios samples) for detection of β-thalassemia using whole gene sequencing and RT-PCR assays. We will share few interesting case studies like co-inheritance of sickle cell anemia and β-thalassemia traits, compound heterozygosity of beta thalassemia major and normal in the case of twin pregnancy. Prenatal diagnosis helps the parents to know the thalassemic status of the fetus in the first trimester screening.
Snigdha Rao
Institute of medical research, Chandigarh, India
Title: Recurrent fetal hydrocephalus: two rare cases of non-familial hydrocephalus recurring in consecutive pregnancies

Biography:
Snigdha Rao is a final year post-graduate student of the prestigious Post-Graduate Institute of Medical Research, Chandigarh, India. She was the best out-going student of her batch in her MBBS from the prestigious Osmania Medical College, Hyderabad. She was the Joint Secretary of OSMECON-12, Undergraduate National Conference and the Editor-in-chief of the conference magazine. She is interested in pursuing fetal medicine.
Abstract:
Hydrocephalus is one of the most common major congenital anomalies occurring in approximately 0.3 to 1/1000 live births. The etiologies of congenital hydrocephalus include infections, vascular abnormalities, mechanical obstruction and chromosomal abnormalities. In genetic terms, the isolated (non-syndromic) form of hydrocephalus is a primary and major phenotype caused by a specific faulty gene. It is estimated that about 40% of hydrocephalus cases have a possible genetic etiology. It can be X-linked or autosomal recessive or even autosomal dominant. The recurrence risk excluding X-linked hydrocephalus is low. Empiric risk rates range from <1% to 4%. Here we present two cases of non-consanguineous couples with no previous family history presenting to us with fetal hydrocephalus in consecutive pregnancies associated with aqueductal stenosis. In our first case the mother had one neonatal loss due to hydrocephalus, termination of second pregnancy due to same defect and a third live birth with the same defect. In our second case the mother had one neonatal loss due to hydrocephalus, termination of second pregnancy for the same and she came to us for pre-conceptional counseling. Genetic analysis was not done in either case. The correct molecular diagnosis can provide the parents with the recurrence risk together with the possibility of prenatal genetic diagnosis for a future pregnancy.

Biography:
Dr. Abdulkarim had completed his postgraduate training in radiology in Leicester training scheme UK where he obtained the FRCR. Currently he is a Consultant Radiologist at George Eliot Hospital and a visiting fellow to Warwick medical school. Dr Abdulkarim’s current research interest is in the field of reduction of intravenous contrast in CT examination and the effects on renal function where he had published several papers.
Abstract:
Polyorchidism is a rare congenital anomaly described as the presence of more than two testes. The aetiology of this condition is unknown but it was first described by Blasius in 1670 as an incidental finding during an autopsy. This is an uncommon condition particularly with an undescended testis and usually diagnosed at early life. We present a case report of 81 year old with undescended supernumerary testis and to our knowledge this is the oldest age at presentation. We will also discuss the relevant embryological development.
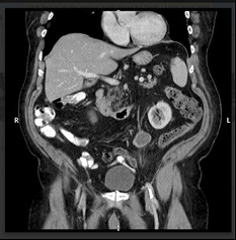
Fig 1: coronal reformat CT image of the abdomen depicting the undescended third testis (seen inferomedial to the left kidney).
References:
- Amodio, John B., et al. "Polyorchidism report of 3 cases and review of the literature." Journal of ultrasound in medicine 23.7 (2004): 951-957.
- Artul, Suheil, and George Habib. "Polyorchidism: two case reports and a review of the literature." Journal of medical case reports 8.1 (2014): 464.
- Avargues, Ana, et al. "Polyorchidism: the case in a young male and review of the literature." Asian journal of andrology 17.3 (2015): 511.
- Bozgeyik, Zülkif, Ercan Kocakoç, and T. Oztürk. "Polyorchidism with lobulation and septa in supernumerary testis." Diagn Interv Radiol 14.2 (2008): 100-2.
- Deveci, Serkan, et al. "Bilateral double testis: Evaluation by magnetic resonance imaging." International journal of urology 11.9 (2004): 813-815.
- Track 13: Endocrinology Case Reports
Location: Duai, UAE

Chair
Jamal Abdulkarim
Dr.

Biography:
Dr. Abdulkarim had completed his postgraduate training in radiology in Leicester training scheme UK where he obtained the FRCR. Currently he is a Consultant Radiologist at George Eliot Hospital and a visiting fellow to Warwick medical school. Dr Abdulkarim’s current research interest is in the field of reduction of intravenous contrast in CT examination and the effects on renal function where he had published several papers.
Abstract:
Recognition of pancreatic anomalies on imaging is essential as they may be of clinical relevance and are potential causes of recurrent pancreatitis or gastric outlet obstruction in patients. (1)
We describe a ‘V shaped pancreas’, a pancreatic anomaly that resembles canine pancreatic anatomy that, to the best of our knowledge, has not previously been described in humans.
We will also review pancreatic embryological development and anatomy, including common variants.
Fig 1: T2 weighted turbo spin echo single shot coronal sequence showing 2 separate moieties of pancreatic tissue separated by fat.